

ͨ�^ꎘO늽��|�fͬ���Ì����늳������H�����l��
���������c��
1. ���������ߌ���������е������s�����O�c�߶ȷ�����(HFE)��늽��|��Y�ϣ�ͬ�r��Q�-��늳������O��ؓ�O���ڵĆ��}��
2. �о���������ʹ��5.8 mg cm-2�ĸ���eSe/Sؓ�d�£�늳���Ȼ�����ṩ> 5.4 mAh cm-2�ĸ���e�������ڻ���HFE��늽�Һ�Ў��Ч��> 99.2%�����⣬�ڸߜأ�55 �棩�͵͜أ�0 �棩�£���Se/S���O��> 5.0 mg cm-2��Ҳ���F��������ѭ�h���ܣ���C��δ���ڌ������ضȷ����ȵČ��H���Ý�����
3. ����߀ͨ�^ͬ��������X-�侀̽ᘺ��w�Еr�g�����x���|�V(ToF-SIMS)Ԕ����ʾ�˝��ڙC�ơ�
���������
������Փ�����ܶȸߣ�2600 Wh kg-1�����-��늳ر��V���J������һ������늳�����ϣ���ĺ��x��֮һ�����ǣ��ڌ��H�����������O������늻��W����߀ԭ�����W��늽��|�Ĵ���ʹ�ã��Mһ���������-��늳ص�ѭ�h�����͌��H�����ܶȡ�
�ڱ����У����߈����һ�N�fͬ���ԣ�ͨ�^������Se/S������O��HFE��늽��|��Y�ϣ�ͬ�r��Q�����ﴩ�����O����߀ԭ�����W�������-��늳��ڌ��H�����l���µ�䇄��x/�僲������}��
������Ԕ�顿
1. OMSH-Se/S���O���ϵĽY���ͽM�ɷ���

�D1. ���O���ϵĽY���ͽM�ɷ���
��D1a��ʾ�������������w��OMSH����SEM�D�����Կ���OMSH�ǿ���~180 nm�������Y����Ȼ������ͨ�^���ڔUɢ����155 ��ض���~ 80 wt.% Se���s����B��OMSH���п����С��õ������O����ӛ��OMSH-Se/S��OMSH-Se/S��TEM�D�D1b���@ʾOMSH�Ĵ�׳�M��Se���s�����w����OMSHSe/S��EDS�D�D1c������S��SeԪ�ؾ���ֲ������O���σȲ����C����Se/S�ɹ����b������Ĵ�Y���С�ͬ�r��Zn��CoԪ�صľ���ֲ�����OMSH-Se/S�Ȳ������p�˽Y��λ�c��������������߀ԭ��늻��W�����W��OMSH-Se/S��XPS�������D1d����ʾ��Se�I�Ĵ��ڣ������R�e��Se 3p1/2 (168.31 eV)��Se 3p3/2 (161.82 eV)�I��ͬ�r��OMSH-Se/S���O�����е�Se-S (56.0 eV & 57.0 eV)��Se-Se (55.49 eV & 56.35 eV)�I���D1e���C������֮���γ��������Se/S������c��S����ȣ�Se/S����γɿ���������O�ͺϲ��ϵ���ӌ���ԺͿ����ԡ�OMSH-Se/S�е�Se/S�������y����79.11 wt. %���D1f����
2. OMSH-Se/S���O���ϵ�늻��W����

�D2. OMSH-Se/S���O���ϵ�늻��W����
HFE��늽��|������Ч���ƶ�������ܽ⣬�Ķ����ƴ���Ч����ͬ�r����ʹ�ڸ������l���£������s������Ĵ�����w�Y��Ҳ���ԘO���������O�ͺϲ��ϵČ���Ժ��x�ӂ�ݔ��������D2a��ʾ��OMSH-Se/S���O���Ͽ�����160 mA g-1������ܶ����ṩ1065.25 mAh g-1�Ŀ���������ߎ��Ч�ʣ�>99.5 %�������⣬��2 mg cm-2�����ܶ��£��Ɍ��F300��ѭ�h��81.77%�ĸ����������ʡ��@�Mһ���C����OMSH-Se/S���O��HFE��늽��|�����õı������ܡ����⣬���Mһ����Se/Sؓ�d���ӵ�5.8 mg cm-2�r�����O��200 mA g-1���Կ��ṩ5.47 mAh cm-2�ı��������D2b��2c����OMSH Se/S�ڸ�Se/Sؓ�d�µĸ��Բ��������ʱ��������������w�cSe���s��Y�Ͽ�����Ч����늻��W����߀ԭ�����W�����⣬���ڶ������ڳ�ҎDME��늽��|�еĸ��ܽ�ȣ����ص��Է�늕�����䇽���ؓ�O����������˥�p���g����D2d��ʾ���ڂ��y�Ļ���DME��늽�Һ�У��o��1�ܺ�늳�늉��½���205 mV������HFE��늽��|��늳��У�늳����o��1�ܺ�늉��H�½���20 mV���D2e��������HFE��늽�Һ�и�С��늉����Mһ���C���˻���HFE��늽�Һ������Ч�ؽ��Ͷ�������Է���ʡ�����Ҫ���ǣ���ʹ���o��1�ܺ��֏�ѭ�h�yԇ�r��늳ص��������������ֵ�5.53 mAh cm-2������167.6 mA g-1��ѭ�h50�κ���5.44 mAh cm-2(�D2f)��
3. ѭ�h늘O�����g���ΑB����
���������˽�ѭ�hSe/S���O��䇽���ؓ�O�ڲ�ͬ늽��|�еĽ�����ΑB���������Mһ���M����ToF-SIMS������늳طքe�ڻ���HFE��DME��늽��|����0.1 C�ij��/��������M��100��ѭ�h���ռ����O��ؓ�O�M�б������ڻ���DME��늽�Һ�У������γɸ߶ȿ��ܵĶ������(LiPSes)�����L��ѭ�h�^���з����ܽ�/�ٳ��e�^�̕�����Se�����O�Ȳ��w�Ƶ�����棬�Ķ���������ѭ�hꎘO�ϵķֲ������D3a����

�D3. ѭ�h늘O�����g���ΑB����
��ʹ�û���HFE��늽��|����r�£��D3b������ѭ�h���O�ı����ϛ]�аl�F���F���w���cSe��ƣ�����DME��늽�Һ��ѭ�h���O��S��̖Ҳ���F���Ȼ���HFE��늽�Һ�е�ѭ�h���O�����������̖���D3a��3b�����Mһ���C���˻���HFE��늽�Һ�ڷ�ֹ���������Ч�ԡ����⣬DME��늽��|��ѭ�hOMSH-Se/S���O��������棨Se-�����D3c�������������F�����ҵ�Se-��̖�������S����ȵ����Ӷ�׃�����C����ѭ�h���O�ı���Se�w�ƌӵ��γɡ���HFE��늽��|����r�£�Se-��̖�ʷ���څ�ݣ�����������/�������ĔUɢ�õ���Ч���⡣�D3d�@ʾ���ڻ���DME��늽�Һ��ѭ�h���䇽���ؓ�O�����Կ���������Ͼ��д����F�K�IJ�ƽ�����棬�����Ǿ����䇄��x/���^�̺͇��ص�䇽��ٸ��g������HFE��늽��|����r�£��D3f���������^�쵽�]�ЈF�K�ľ�����棬����HFE��늽��|�е��Ó��/���e�^�̷�����䇽��ٸ��g���ޡ���D3e��ʾ���ڻ���DME��늽��|�У�������ѭ�h��䇽���ؓ�O����z�y��Se-��̖���@�����������w�Ƶ�ؓ�O�Ȳ�ֱ���c䇽���ؓ�O���������֮�£�ʹ�û���HFE��늽��|��ѭ�h䇽���ؓ�O�ı����ϟo���z�y����Ҋ��Se-���D3g�������⣬����Li-N��Li-F�����ﱻ�J����䇽���ؓ�O����SEI����Ҫ�ɷ֣������x��N-��F-��̖����ָ�ˁ������ɷN��ͬ늽��|���γɵ�SEI����D3e��ʾ��N-��F-��̖�ڂ��y�Ļ���DME��늽��|�зֲ����������γɵ�SEI��ѭ�h�^���к��y�������Π�ڻ���HFE��늽��|�У�N-��F-��̖�ֲ����D3g����������䇽���ؓ�O�����γ��˷�����SEI��
4. ���O���ϵ�ԭλ��׃����
���ߌ�OMSH-Se/S���O��HFE��늽��|�г��/������g�M��ԭλ����X�侀����(HEXRD)�������ԱO�y��׃����D4a��ʾ��λ��4.48°��6.39°��6.71°��7.48°��7.75°��8.03°��8.33°��8.39°̎�ķ���Է���o�Y��Se���sS�࣬�䏊���S�������ȵ������_ʼ���ͣ��f������^����Se/S���O����߀ԭ������Se���s���c�˜ʽY��S��ȣ�ԭʼOMSH-Se/S���O�ķ�λ�����p��׃������~1.5 V�ķ�늽�ֹ늉��£�һЩ������S�����ʧ�ˣ�4.48°��7.2°��8.33°��8.39° �� 9.9°������һЩ�壨7.5°��7.8° , �� 9.1˚)��Ȼ���ڣ����c�˜�Li2Se��Li2S�ķ��دB���M����ˣ����Կ���7.8˚̎�ķ及�ȏ�3.33С�r���ӵ�4.8С�r���@��ԓ������Li2S���γɡ����⣬���]��늳���ԭλXRD�y���^���еĸ߷�늱�����(1206 mAh g-1)���֓�����S�ڷ���^���б�߀ԭ�γ�Li2S��Li2Se��ͬ�r��λ��10.43˚�ķ及����~1.0 h�_ʼ���ӣ�������u�γ���һ�N�Y�����g�w��֮��ԓ��ֵ�����ڽӽ�����^�̽Y���r�_ʼ���ͣ����ڽ�ֹ늉�̎����ʧ���ڳ���^���У�10.43˚�及���ڼs7.0 h�_ʼ���ӣ�Ȼ��Ѹ����ʧ�������ڳ���^����Ҳ�����@�N�Y�����g�w������֮ǰ��ԭλXRD��Li2S2����Փ�о���ԓ�������Li2S2���Mһ���ij���^��������̖�Ļ֏ͣ������ڳ���^����Li2S��Li2Se�������D����Se���s��S��ԭλHEXRD�Y���C����OMSH-Se/S���O��HFE��늽��|�о��Ѓ����Ŀ����Ժ��Բ�������Ч�ʡ�
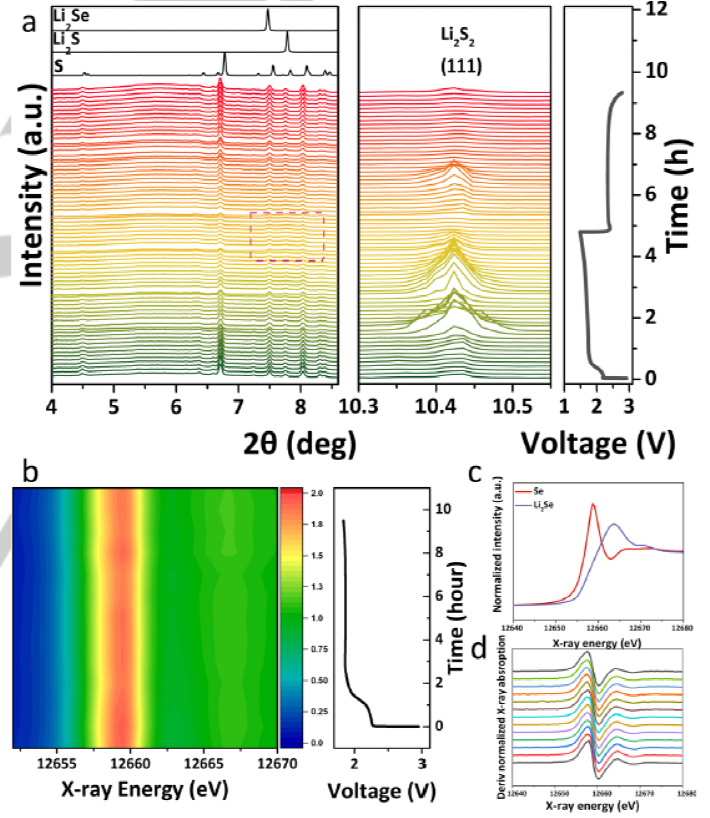
�D4. ���O���ϵ�ԭλ��׃����
�������ý�߅���Y��ԭλX�侀����(XANES)�Mһ����̽�y�ڷ����1.8 V�^����Seԭ���܇��ĭh���M���˱�������D4b��4c��ʾ���S�������ȵ����ӣ�Se���������ȣ�~12658.7 eV����u���ͣ�����Se/S���B�m߀ԭ��ͬ�r��Li2Se���������ȣ�~12663.6 eV���ڷ���^������u���ӣ������ڷ���^�����γ���Li2Se�a����⣬��D4b��4d��ʾ��Seԭ����12658.7 eV �� 12663. 6 eV֮�g�]�������D�ƣ��@�c֮ǰ�����Se/S���O��̼���}��늽��|�е��О�һ�£����cDME��늽��|�е��О鲻ͬ����ˣ����Եó��YՓ��HFE��늽��|�е�OMSH-Se/S���O���v�˜ʹ�-�̣�Ó��䇻��^�̣��@�^�˶��������g�w���γɣ��Ķ������˴������������˥�p��
5. OMSH-Se/S���O�cHFE��늽��|�M�ϵČ��H������C
��������������Ĵ�����w���Ϻ�Se���s����Ч�������OMSH-Se/S���O��늻��W����߀ԭ�����W����D5a��ʾ������OMSH-Se/S���O(5.2 mg cm-2)��0 ����ѭ�h�r�����O��100��ѭ�h���Կ��ṩ������> 4.2 mAh cm-2�Ŀ���������Ȼ�����ؗ͜l����Ӱ�늻��W����߀ԭ��SEI�γɄ����W�����⣬늽��|���x��늌��ʵ��������|Ҳ���ܵ��ؗ͜l����Ӱ푡���ˣ�SEI�ڵؗ͜l�����γɺͷ�����Ҫ���L�ĕr�g����������ѭ�h���ܮ�����Ȼ����һ��SEI�ڎׂ�ѭ�h���γ����ã�ѭ�h���ܾͿ��Ի֏�������Ȼ�������u���˺�OMSH Se/S���O��5.0 mg cm-2����HFE��늽��|��55��ĸߜ��µ�늻��W���ܡ�

�D5. OMSH-Se/S���O�cHFE��늽��|�M�ϵČ��H������C
��D5b��ʾ���^�ߵĜضȿ����Mһ������OMSH-Se/S���O��늻��W����߀ԭ�����W���Ķ��a��1215.97 mAh g-1�ĸ߱����������ҿ��Ա���750.94 mAh g-1�ı���������ʹ�ڸ���eSe/Sؓ�d�£�50��ѭ�h�����ܱ���750.94 mAh g-1�ı����������ڸߜؗl�������¾۱�ϩ��Ĥ�ğ�׃�Σ�����߀�����������������W�����������ѭ�h��Ҳ�^�쵽�˷����^�̡�Ȼ�������^�ׂ�ѭ�h��һ���Ȳ��h�������������ܻ֏�������
�����S��ʹ�Ì��õ�ܛ��늳����Üyԇ��OMSH-Se/S���O��HFE��늽��|�е�늻��W���ܡ�ܛ��늳�������D5c��ʾ�����������u���˻���OMSH-Se/S���O��HFE��늽��|��Li-Se/Sܛ��늳ص����ܣ���200 μm���䇽��ٲ���ؓ�O������E/S�ȿ�����10 μL mg-1����D5d��5e��ʾ���M�b���Li-Se/Sܛ��늳���200 mA g-1������ܶ��¿��ṩ830.76 mAh g-1�ij�ʼ��������ͬ�r��50��ѭ�h�����ܺܺõر���650.23 mAh g-1�ı����������������������ʞ�78.27%��
�����ṩ���挍��늳ر������������J��߀���м�����䇽���ؓ�O��늽��|��������ˣ����ߑ��ñ�䇽��ٲ���40 μm������ؓ�O������Li-Se/Sܛ��늳ص�E/S�ȿ�����7 μL mg-1���Ķ�����Li-Se/Sܛ��늳صķ����ԡ���D5f��5g��ʾ���M�b���Li-Se/Sܛ��늳���200 mA g-1�r���ṩ931.12 mAh g-1�ij�ʼ�����������⣬ԓ늳���70��ѭ�h�п��Ժܺõر���> 99.3 %�ĸߎ��Ч�ʡ�
Chen Zhao, Amine Daali, Inhui Hwang, Tianyi Li, Xingkang Huang, David Robertson, Zhenzhen Yang, Steve Trask, Wenqian Xu, Cheng-Jun Sun, Gui-Liang Xu, Khalil Amine, Pushing lithium-sulfur batteries towards practical working conditions through cathode-electrolyte synergy, Angewandte Chemie International Edition, 2022, https://doi.org/10.1002/anie.202203466




�����Wע�� ����Դ��XXX�����Ї�늳��ˣ�������Ʒ�����D�d������ý�w���D�dĿ�����ڂ��f������Ϣ�������������Wٝͬ���^�c�͌����挍��ؓ؟��
������Ʒ���ݡ������������}��Ҫͬ���Wϵ�ģ�Ո��һ�܃��M�У��Ա��҂����r̎����
QQ��503204601
�]�䣺cbcu@www.69gh.com
-
������ˮ���z늽��|���� �����_�l�ɳ��ˮϵ�\�x��늳�
2022-02-23 10:38 -
�о��ˆTͨ�^�����OӋ������늳؉��� ʹ늄���܇�m������ȼ��܇
2022-02-12 14:48 -
�µ��|�ӂ���늽��|�OӋԭ���������Мع̑B������ȼ��늳�
2022-01-03 13:45 -
��헿Ƽ���W�_�l�{�Y��늽��| ��߹̑B늳ص��x��늌���
2021-08-24 08:44 -
99�q�늳�֮������Փ�ģ��ٴξ۽��̑B늽��|��
2021-07-20 11:34 -
�������������Ƽ���W��ͨ�^���ƶ����ﴩ�� ������늳�����
2021-07-02 22:03 -
���̎��_�l���ɷ�����B늽��|�ĸ�Ĥ ʹ���͜�늳ظ���ȫ
2021-06-10 08:45 -
�о��ˆT�_�l����늽��| ����䇿՚�늳�ѭ�h������
2021-05-14 08:50 -
�����̑B늳��аl���g�Mչ���{���w�S�W�j�ۺ���ͺ�늽��|
2021-05-06 08:45 -
MIT�l�F��늽��| 늳������ܶȻ��_��420�ߕr/����
2021-04-01 11:21
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



-
������ˮ���z늽��|���� �����_�l�ɳ��ˮϵ�\�x��늳�
2022-02-23 10:38 -
�о��ˆTͨ�^�����OӋ������늳؉��� ʹ늄���܇�m������ȼ��܇
2022-02-12 14:48 -
�µ��|�ӂ���늽��|�OӋԭ���������Мع̑B������ȼ��늳�
2022-01-03 13:45 -
��헿Ƽ���W�_�l�{�Y��늽��| ��߹̑B늳ص��x��늌���
2021-08-24 08:44 -
99�q�늳�֮������Փ�ģ��ٴξ۽��̑B늽��|��
2021-07-20 11:34 -
�������������Ƽ���W��ͨ�^���ƶ����ﴩ�� ������늳�����
2021-07-02 22:03 -
���̎��_�l���ɷ�����B늽��|�ĸ�Ĥ ʹ���͜�늳ظ���ȫ
2021-06-10 08:45 -
�о��ˆT�_�l����늽��| ����䇿՚�늳�ѭ�h������
2021-05-14 08:50
-
���U��늳خaƷ̼���E�u�r���t���˜ʌ�����������_
2022-04-08 10:27 -
�}����䇸߾��� �@����I�٫@��Σ�
2022-04-07 17:26 -
���늳���I��� ���ؓ�O�F����Σ�
2022-04-08 18:11 -
��������Դ�惦�ش�ͻ�ƣ����͡�����늳ء��H���ܟ�rጷ�����
2022-04-08 11:43 -
��늳��t���������x�����˳��L��ͬ�������\ϵ��һ��
2022-04-06 08:45 -
�ȁ��ϡ���˹�������r�������̛_��IPO ��������Դ����������|��
2022-04-08 11:19 -
�о��ˆT����X�侀���@EV늳ص��˻��D��
2022-04-08 08:58 -
���r���D�y�������ˡ�
2022-04-11 11:21

�rֵ�ɾ��ИIƷ�ƣ����\�����ṩ���������YӍ
��ICP��09081210̖
 ��I��̖
��I��̖ �Ź���̖
�Ź���̖